AG Eiseler: Exosomes in Metastasis and Inflammation
Projects
Pancreas adenocarcinoma and small extracellular vesicles
Pancreatic ductal adenocarcinoma (PDAC) are characterized by a dismal prognosis due to late-stage diagnosis and early metastasis1. It is therefore vital to understand which factors determine PDAC metastasis patterns. In the last years it has become more and more evident that small extracellular vesicles (sEVs, exosomes) that are released by tumor cells in high amounts are vital regulators of tumor progression and metastasis2-4. sEVs have a size range of 30-150 nm and are derived of the endo-lysosomal system. sEV originate in multivesicular bodies (MVBs) as intraluminal vesicles. Once sEVs have been released by tumors cells, they can be distributed via the circulation. Recipient cells are educated by binding of sEVs to surface receptors or intracellular release of their bioactive cargos, such as lipids, proteins, miRNAs or mRNA thereby modifying the behavior of cells2,4.
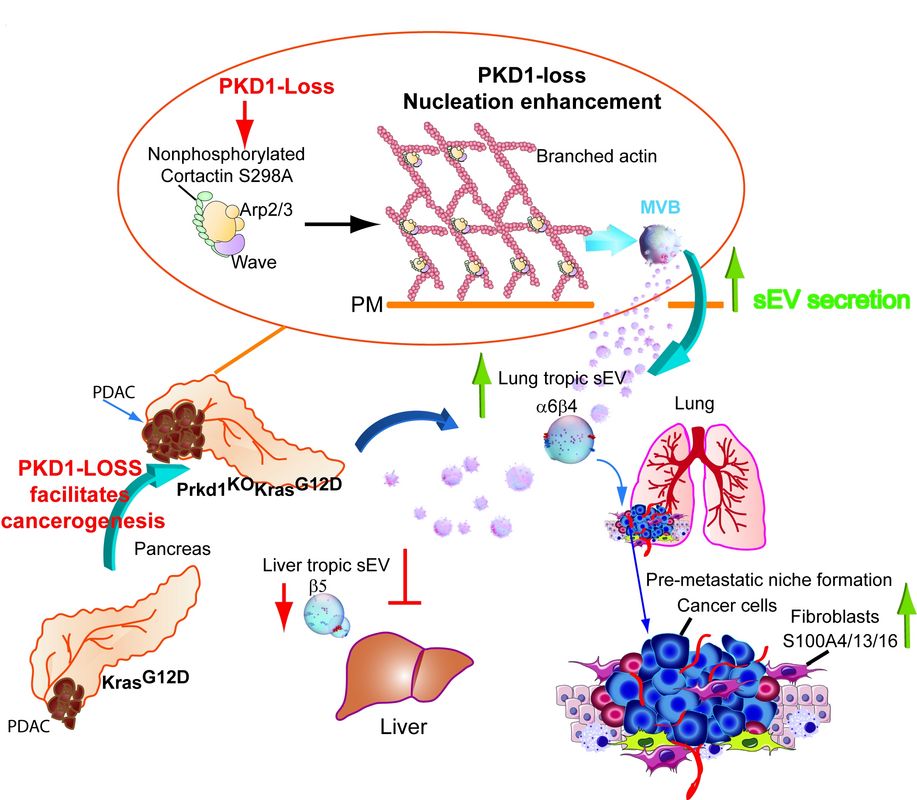
a. Intercellular communication during PDAC invasion and metastasis
Recently it was shown that distinct integrin combinations on the surface of sEVs from cancer cells are able to direct organotropic metastasis5,6. This is achieved by preferred uptake of the tumor-derived sEVs by cells in future metastatic organs e.g. fibroblasts in the lung6. The sEVs carry bioactive molecules which change the behavior of recipient cells to support the establishment of pre-metastatic niches6. Our group has recently shown that Protein Kinase D1 (PKD1) is downregulated in 72% of PDAC by more the 75%7. Loss of PKD1 strongly enhanced sEV secretion and directed metastasis of PDACs to the lung in a Prkd1KOKC mouse model. We demonstrate that this was facilitated by upregulation of integrins a6b4 in cells and on sEVs. These integrins are packaged into Prkd1KO-derived sEVs in a CD82-dependent manner7, by facilitating their recycling from the cell surface into MVBs. We also demonstrate that strongly increased sEV secretion upon loss of PKD1 expression is achieved via the PKD1 substrate Cortactin. When Cortactin is not phosphorylated by PKD1 at S298, it is able to promote the formation of branched actin filaments at the plasma membrane which are required for the efficient fusion of MVBs and thus sEV release (Fig. 1). Once these sEVs have reached the lung, we show uptake into lung fibroblasts facilitates the formation of pre-metastatic niches via S100A proteins6,7. As part of project C1 GRK2254 “Heterogeneity and Evolution in Solid Tumors (HEIST)”, we are currently investigating how sEVs released by PDACs define pre-metastatic niches in the lung or liver microenvironment on a physiological and molecular level. Besides, we are elucidating how relevant cargos are packaged into sEVs and if this process can be targeted to qualitatively alter sEV cargo content and distant metastasis.
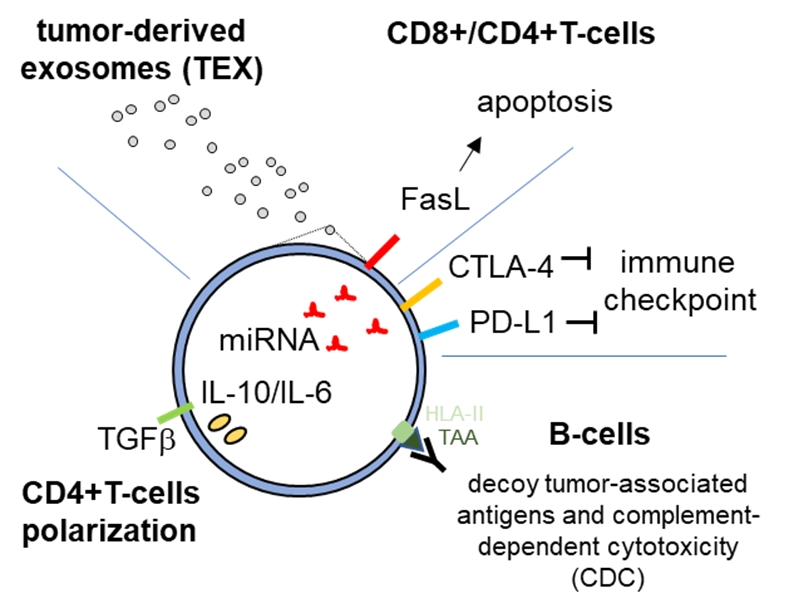
b. sEV mediated immune evasion in PDAC
The PDAC microenvironment is highly immunosuppressive, blocking CD8+ T-cell tumor recognition and clearance8,9. sEVs are associated with different human diseases, including cancer, as levels of plasma sEVs were shown to be extensively enhanced in cancers patients. Tumor-derived sEVs were shown to carry immune-inhibitory ligands such as PD-L1, FasL and CTLA-4 that can directly interact with receptors on target cells, interfering with immune cell function10-14. We are currently analyzing how immune-modulatory ligands are packaged into sEVs and how these factors modulate T-cell immune evasion via CD8+ and CD4+ T-cells, contributing to the highly immunosuppressive PDAC tumor microenvironment. To this end, we are characterizing sEV miRNA cargos and protein targets for functions in apoptosis, proliferation, T-cell polarization and activation. Experiments are performed in vitro and in PDAC mouse models.
Physical trauma
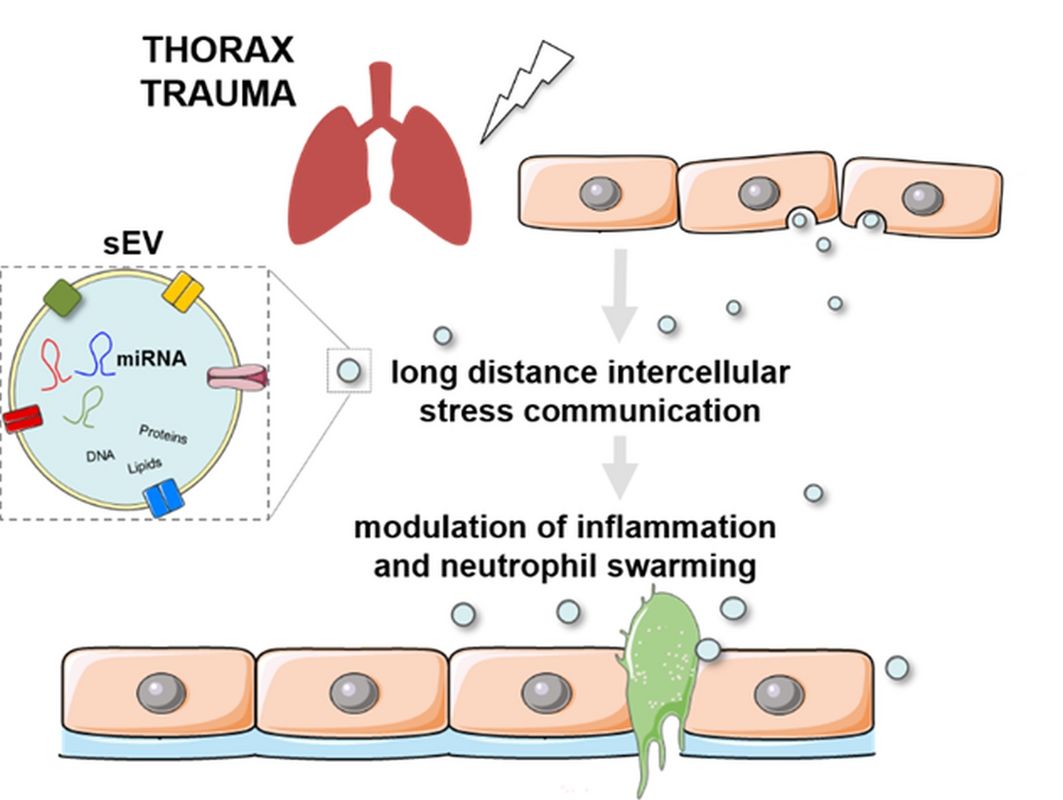
a. Role of sEVs in the host inflammatory trauma response
Trauma is the leading cause of death under 44 years of age15,16. A traumatic event triggers the release of damage- and pathogen-associated molecular patterns (DAMPS, PAMPs) causing neutrophil-mediated inflammation and barrier disruption17-20. Severe injuries in combination with comorbidities can also cause a runaway systemic inflammatory response that can even manifests in multiple-organ-dysfunction (MODS)21. In particular thorax trauma (TxT) is associated with one quarter of all trauma-related deaths22. Thus, it is important to understand how such an inflammatory response to a physical trauma is coordinated and whether it can be mitigated. Small extracellular vesicles (sEVs, exosomes) were described to be important mediators of stress signalling and are involved in immune modulatory processes4,12. However, molecular mechanisms and functions of sEVs after a traumatic insult have not been elucidated in detail. As part of the CRC 1149 “Danger Response, Disturbance Factors and Regenerative Potential after Acute Trauma” we are currently investigating whether qualitative and qualitative changes in sEVs after a traumatic insult will be implicated in propagating local, distant and systemic inflammatory processes.
b. Regulation of neutrophil chemotaxis and endothelial barrier stability
Neutrophils are important mediators of the innate immune defense and of the host response to a physical trauma23. We have previously reported that inhibition of Protein Kinase D by the small molecule inhibitor CRT0066101 was able to inhibit transendothelial migration of neutrophils towards a chemotactic gradient. To this end, PKD phosphorylates the Cofilin-phosphatase Slingshot-2L (SSH-2L). SSH-2L in turn dynamically regulates Cofilin activity and actin polymerization in response to a neutrophil chemotactic stimulus, such as fMLP23. Impaired PKD activity also significantly reduced neutrophil deformability as determined by optical stretcher analysis. Indeed, we were able to inhibit transmigration (diapedesis) of neutrophil-like differentiated NB4 cells and primary PMNs23. Thus, our data suggest that inhibition of PKD may affect trauma outcome by directly acting on neutrophil transmigration. In addition, we have elucidated a molecular mechanism by which inhibition of PKD activity is implicated in stabilizing epithelial24 and potentially also endothelial barriers via its substrate Cortactin24. We are therefore currently investigating in a DFG funded project, whether inhibition of PKD will improve outcome after thorax trauma (TxT) and mitigate a neutrophil-driven inflammatory response in vivo.
PKD family of serine/threonine kinases
PKD kinases are also known to drive vesicle fission from the trans-Golgi network25. We are investigating how specific PKD isoforms, in particular PKD2, regulate and execute different steps of vesicle budding and fission by a network of protein interactions involving the small GTPase Arf1 26. Also, regulation of Cortactin and actin polymerization during vesicle separation from Golgi membranes27 and fusion of MVBs with the plasma membrane is investigated7. In order to address these questions, we are studying compartment-specific protein dynamics, interactions and activation processes utilizing high-end confocal microscopy techniques, such as Fluorescence-Recovery-after-Photobleaching (FRAP) and Foerster Energy Transfer (FRET) complemented by biochemical assays.
Cooperation partners
- Prof. Dr. Johan Van Lint, Katholieke Universiteit Leuven, Belgium
- Prof. Dr. med. Alexander Kleger, Institute of Molecular Oncology and Stem Cell Biology, University of Ulm
- Prof. Dr. Franz Oswald, Department for Internal Medicine I, University of Ulm
- Prof. Dr. Mechthild Haztfeld, Division of Pathobiochemistry, Martin-Luther University, Halle
- Dr. Martin Müller, Center for Internal Medicine I, University of Ulm
- Dr. Angelika Rück, Core Facility for Confocal and Multiphoton Microscopy
- Dr. Stephan Paschke, Department of Visceral Surgery, University of Ulm
- Prof. Dr. Ralf Kemkemer, Max Planck Institute for Intelligent Systems, Stuttgart/University Reutlingen
- Dr. Julia von Blume, Max Planck Institute for Biochemistry, Munich