Anämie
Ansprechpartner
Hämatologische Fachsprechstunde
Anmeldung zur Spezialsprechstunde unter Infos für Ärzte
Bei Patienten mit chronischen Krankheitsformen die erstmals zu uns kommen ist es sinnvoll, wenn wir im Vorfeld die wichtigsten Befunde und Berichte erhalten, um die Problematik in der Anämiekonferenz besprechen und interne Funktionsuntersuchungen und Beratungstermine zu planen. Dasselbe gilt für Patienten deren Wohnort soweit entfernt ist, dass sie stationär genommen werden oder in Ulm übernachten müssen.
Klinische Studien
Weitere Informationen zu den aktiven Studien erhalten Sie unter:
Diagnostik und Therapie in der Klinik für Innere Medizin III
Anämien gehören zu den häufigsten Folgeerscheinungen zahlreicher Allgemeinerkrankungen, vor allem bei infektiösen und nichtinfektiösen Entzündungen, Mangelzuständen, beim Nierenversagen und bei Neoplasien. Die Diagnostik und ursachenorientierte Behandlung erfolgt in der Mehrzahl der Patienten in der Allgemeinpraxis, beim niedergelassenen Internisten oder Kinderarzt. Die Mitwirkung des Facharztes für Hämatologie in der Praxis oder der Klinik ist allerdings bei einem Teil der Betroffenen erforderlich. Dazu gehört die Diagnostik bei Patienten mit ungeklärter Anämie sowie die Therapie und Verlaufskontrolle bei seltenen Formen wie den autoimmunhämolytischen Anämien, und bei komplexen Bluterkrankungen (Leukämie, Myelodysplastisches Syndrom, aplastische Anämie).
Ein besonderes Problem bieten Patienten mit angeborenen Anämien, ganz überwiegend aufgrund eines weiten Spektrums genetischer Defekte. Sie werden oft schon im Kindesalter diagnostiziert und sind meist über Jahre in kinderärztlicher Betreuung. Durch die Fortschritte in der Behandlung hat sich die Lebenserwartung von Patienten mit angeborenen Anämien erheblich verbessert, sodass zunehmend internistische Folgezustände und Komplikationen zu beherrschen sind. Dies erfordert eine umfassende interdisziplinäre Betreuung auch im Erwachsenenalter. Die Häufigkeit von Patienten mit erblichen Hämoglobinopathien wie Thalassämien oder Sichelzellerkrankung sind aufgrund der Immigration aus Endemiegebieten weit häufiger als früher. Nur eine aufgrund der Erkenntnisse aus diesen Ländern mögliche spezialisierte Betreuung erlaubt eine fachgerechte Intervention in Krisensitutationen und eine Verbesserung der Lebensqualität und Normalisierung der Lebenserwartung. Im Gegensatz zu den Mittelmeerländern und USA, Großbritannien und Frankreich, die schon seit Jahrzehnten spezielle Zentren zur Behandlung dieser Patienten eingerichtet haben, existieren in Deutschland bislang keine Strukturen, welche die lebenslange Betreuung erwachsener Patienten und die Beratung der am Wohnort verantwortlicher Ärztinnen und Ärzte sicherstellen.
In enger Kooperation mit den Kollegen der Klinik für Kinder- und Jugendmedizin und der Abteilung für Transfusionsmedizin soll die Diagnostik und Behandlung seltener erworbener wie angeborener Anämien bei erwachsenen Patienten in der Klinik für Innere Medizin III zentralisiert und verbessert werden. Ein Beispiel dafür ist das seit über 20 Jahren bestehende Register zur Dokumentation und Beratung von Patienten mit Congenitaler Dyserythropoetischer Anämie (CDA), geführt von Herrn Prof. Dr. med emerit. H. Heimpel (Klinik für Innere Medizin III) in Zusammenarbeit mit Frau Prof. Dr. med Kohne (Klinik für Kinder- und Jugendmedizin) und Prof. Dr. med H. Schrezenmeier (Institut für Klinische Transfusionsmedizin und Immungenetik Ulm, IKT). Damit verbunden ist eine kontinuierliche Konsiliartätigkeit und Beratung von ärztlichen Kollegen aus Deutschland und angrenzenden europäischen Ländern.
Laboratoriumsdiagnostik
Blutanalyse: Zellzählung, Erythrozytenindices, morphologisches Blutbild.
Knochenmarkdiagnostik: Zytologie, Eisenfärbung, Flow Zytometrie, Histologie (Prof. Möller), Zytogenetik, Molekulare Diagnostik. Eisenstatus, Haptoglobin, weitere Laborparameter in Zusammenarbeit mit dem Institut für klinische Chemie.
Nicht-invasive Messung des Eisengehalts durch MRT (Klinik für diagnostische und interventionelle Radiologie).
Spezielle Hämolyseparameter, Analyse der Erythrozytenenzyme, Eosin-Malein-Test (EMA-Test), biochemische und genetische Hämoglobinanalyse (Klinik für Kinder–und Jugendmedizin, Prof. Dr. med Kohne).
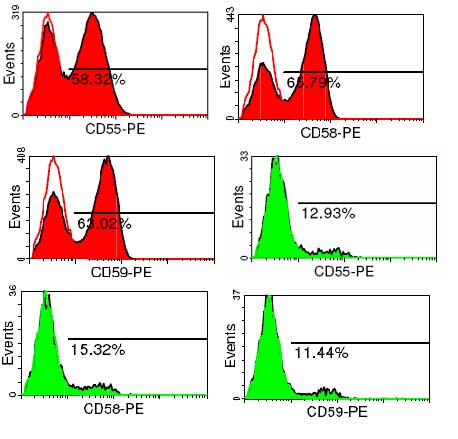
Flow Zytometrie zum Nachweis einer PNH, Immunhämatologie (IKT), genetische Diagnostik bei V.a. auf kongenitale dyserythropoetischen Anämien und bei PNH: Institut für klinische Transfusionsmedizin und Immungenetik (IKT) (Prof. Schrezenmeier, Dr. K. Schwarz).
Beschreibung des Krankheitsbildes und Basisinformation
Von einer Anämie (Blutarmut) spricht man, wenn eine Verminderung der Hämoglobinwerte (Hb) oder des Hämatokrits (Hk) unter einen alters- und geschlechtsspezifischen Referenzwert nachweisbar ist. Anämien sind eine der häufigsten krankhaften Veränderungen weltweit und meist Folge chronisch entzündlicher oder neoplastischer Erkrankungen. Bei sehr vielen Bluterkrankungen tritt eine Anämie als Teilbefund auf. Eine Anämie entsteht entweder durch zu raschen Abbau (Hämolyse) - erworben durch anti-erythrozytäre Autoantikörper oder angeboren durch Enzym- oder Membrandefekte - verminderte (z.B. Aplastische Anämie) oder ineffektive (Myelodysplastisches Syndrom, MDS) Bildung oder eine Verteilungsstörung (Schwangerschaftsanämie, Hypersplenimus) roter Blutkörperchen. Eine verminderte Hämoglobinbildung im Rahmen eines Eisenmangels, Vitaminmangels oder bei angeborenen Anämien kann ebenfalls zu einer Blutarmut führen. Anämien bei chronischen Erkrankungen – auch Tumorerkrankungen - sind meist durch die Kombination mehrerer Ursachen bedingt.
Referenzwerte:
14 - 70 Jahre Männer: Hb 13 – 17 g/dl; Hkt 42 – 50 %; Erythrozyten 4,3 – 5,6 T/l
14 - 60 Jahre Frauen: Hb 12 – 16 g/dl; Hkt 38 – 44 %; Erythrozyten 4,0 – 5,4 T/l
Definition einer Anämie nach WHO:
Männer Hb < 13 g/dl
Frauen Hb < 12 g/dl
Eisenmangelanämie
Eine der häufigsten Anämieformen ist die Eisenmangelanämie. Die Diagnose wird anhand einer charakteristischen Konstellation mit Nachweis einer hypochromen, mikrozytären Anämie mit Verminderung des mittleren kopuskulären Hämoglobingehaltes (MCH) und/oder des mitteren Erythrozytenvolumens (MCV) gestellt. Eine Eisenmangelanämie entsteht durch eine gestörte Balance zwischen Eisenverlust und Eisenaufnahme. Am häufigsten liegt ein anhaltender Blutverlust, seltener eine reduzierte Eisenaufnahme zugrunde. Sie ist keine eigenständige Erkrankung, sondern Symptom einer zugrunde liegenden Störung. Die Ursache des Blutverlustes zu finden ist die Aufgabe des Arztes und sollte vor einer Behandlung mit Eisenpräparaten stehen. Häufige Ursachen sind Blutverluste über den Darm oder den Harntrakt und bei Frauen über die Regelblutung. Ein Eisenmangel kann nachhaltig nur durch Beseitigung der Ursache behandelt werden.
Anämie bei chronischer Erkrankung
Ebenfalls häufig ist die Anämie im Rahmen chronischer Erkrankungen (Anemia of chronic disease; ACD). Sie tritt im Rahmen von Tumorerkrankungen oder chronischen Entzündungsprozessen (Rheumatoide Arthritis, Lupus erythematodes, Tuberkulose) auf. Die ACD ist meist normochrom-normozytär, in einem Drittel der Fälle hypochrom-mikrozytär und dann von einer Eisenmangelanämie abzugrenzen. Das Serumferritin ist in der Regel erhöht. Die Behandlung orientiert sich an der Grunderkrankung.
Anämie bei Mangel an Vitamin B12 oder Folsäure
Als Folge eines Mangels von Vitamin B12 oder Folsäure entsteht eine Megaloblastäre Anämie. Sie ist gekennzeichnet durch eine hyperchrome (MCH > 34) und makrozytäre Anämie (MCV >100). Charakteristisch sind niedrige absolute Retikulozytenzahlen sowie eine häufig stark erhöhte Laktatdehydrogenase (LDH). Die Diagnose wir der Regel durch die klassische Laborkonstellation in Verbindung mit einem niedrigen Vitaminspiegel gestellt. Die Behandlung besteht in der Gabe des fehlenden Vitamins.
Hämolytische Anämien
Ein zu rascher Abbau von Erythrozyten mit Ausbildung einer Anämie wird hämolytische Anämie genannt. Als erworbene Ursachen kommen unter anderem Autoimmunhämolysen mit Nachweis von Antikörpern welche die roten Blutkörperchen besetzen und zerstören sowie seltene erworbene Membranveränderungen (Paroxysmale nächtliche Hämoglobinurie) in Frage. Die Labordiagnostik umfasst die klassischen Hämolyseparameter (LDH, Bilirubin, Haptoglobin) sowie spezielle Diagnostik zum Nachweis von Autoantikörpern (Coombs-Test) und Membrandefekten (FACS-Analyse).
Die Behandlung ist unterschiedlich und richtet sich nach der Ursache der Hämolyse. Autoimmunhämolysen werden primär mit Kortikosteroiden und /oder Immunsuppressiva therapiert. Die PNH kann den Einsatz des monoklonalen Antikörpers Eculizumab erforderlich machen. Patienten mit therapierefraktärer hämoytischer Anämie profitieren manchmal von einer Milzentfernung.
Angeborene Anämien
Thalassämien
In Deutschland sind die Thalassämien nach wie vor selten, allerdings nimmt deren Prävalenz durch Zuwanderung aus dem mediterranen, afrikanischen und asiatischen Raum zu. Die Thalassämie ist die häufigste Form der angeborenen mikrozytären Anämien.
Ursachen
Durch einen genetischen Defekt kommt es zu einer Minderproduktion von Hämoglobinketten. Je nachdem welche der Ketten vermindert gebildet wird, nennt man die Erkrankung alpha-Thalassämie (selten) oder beta-Thalassämie (häufig). Die Überlebenszeit der roten Blutkörperchen wird verkürzt, es kommt zur Hämolyse.
Diagnostik
Eine Verdachtsdiagnose kann meist aus der Konstellation der Blutbildparameter gestellt werden; sie bedarf der Bestätigung durch die Hämoglobinanalyse (beta-Thalassämie) oder einer genetischen Testung.
Krankheitszeichen
Das klinische Bild ist abhängig von der genetischen Konstellation:
Thalassämia minor: Heterozygotie: nur ein Allel (Genkopie) ist mutiert, dies ist meist symptomlos. Die Erythrozyten sind klein (MCV und MCH sind vermindert).
Thalassämia intermedia: Meist compound Heterozygotie: die beiden Allele sind an unterschiedlichen Stellen mutiert, es entsteht eine mittelschwere Anämie mit Ikterus („Gelbsucht“) und Milzvergrößerung. Je nach Ausprägung kann eine Eisenüberladung entstehen.
Thalassämia major: Meist Homozygotie: beide Allele sind identisch mutiert, es entsteht eine schwere, im Kindesalter auftretende schwere Anämie mit zunehmender Milzvergrößerung und Wachstumsverzögerung. Früher war die Haupttodesursache eine Eisenüberladung mit Organschädigung.
Therapeutische Möglichkeiten
Alle leichten Formen bedürfen keiner Behandlung. Die mittelschweren und schweren Formen erfordern bereits im Kindesalter regelmäßige Transfusionen mit daraus resultierender Eisenüberladung (Transfusionshämosiderose). Daher kommt der Kontrolle dieser Komplikation maßgebliche Bedeutung zu. Die Verfügbarkeit von Chelatbildnern hat die Behandlung wesentlich verbessert, insbesondere hat der Einsatz moderner, als Tabletten verfügbarer Medikamente die Behandlung erleichtert. Im Laufe des Lebens kann die Entfernung der Milz erforderlich werden.
Einzig potentiell kurative (auf Heilung ausgerichtete) Behandlung ist bislang die Knochenmark- oder Blutstammzelltransplantation. Wegen der damit verbundenen Gefahren, wird sie lediglich bei schweren Formen eingesetzt.
Qualitative Hämoglobindefekte (Hämoglobinopathien)
Qualitative Hämoglobindefekte (Hämoglobinopathien)
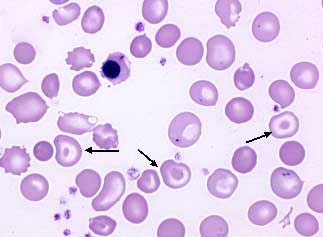
Sichelzellkrankheit
Die Sichelzellkrankheit ist die häufigste Hämoglobinopathie weltweit, in Deutschland leben derzeit über 1000 Patienten, Ausschliesslich Migranten aus Zentral- und Westafrika, den Ländern des östlichen Mittelmeerraumes dem Mittleren Osten, Indien und Zentralasien.
Ursachen
Durch eine Punktmutation an Position 6 der beta-Hämoglobin-Kette (Glu-Val) das Hämoglobinmolekül (HbS) derart verändert, dass im sauerstoffarmen Zustand das Hämoglobin von der löslichen Form in ein Polymer übergeht und die Gestalt der Erythrozyten in die Form einer Sichel bringt. Sichelzellträger bei denen nur ein Allel getroffen ist sind weitgehend symptomfrei, bei Homozgotie oder bei Kombination mit einem Thalassämiegen entsteht die Sichelzellkrankheit.
Diagnostik
Blutausstrich mit Nachweis von Sichelzellen (Abbildung 2: Blutausstrich eines Patienten mit Sichelzellanämie). Hämoglobinanalyse mit Nachweis von HbS und ggf. weiterer H. Molekulargenetische Analyse.
Krankheitszeichen
Die Verformung der Erythrozyten führt zu einer Verstopfung von kleinen und kleinsten Blutgefäßen in inneren Organen (Leber, Milz, Knochen, Gehirn, Netzhaut, Nieren, Lunge etc.). Die Sichelzellkrankheit führt zu Durchblutungsstörungen bis hin zu Infarkten. Folgende Komplikationen sind häufig:
- Aplastische Krise mit schwerer Anämie nach Virusinfektionen (Parvovirus B19)
- Infektionen mit Bakterien (Pneumokokken, Salmonellen)
- Akutes Thorax-Syndrom: Blutansammlung in den Gefäßen der Lunge mit Brustschmerzen, Atemnot, Fieber und Sauerstoffmangel
- Schmerzkrisen vor allem im Rücken, Armen, Beinen, Brust und Bauch.
- Knochenentzündungen (Osteomyelitiden)
- Schlaganfälle, Hirnblutungen
Therapeutische Möglichkeiten
Hydroxyurea senkt bei einem Teil der Patienten die Anzahl und Intensität der Schmerzkrisen und reduziert die Sterblichkeit.
Transfusionen sollten so zurückhaltend wie möglich und lediglich bei Sequestrationskrisen und aplastischen Krisen gegeben werden. Bei schweren Komplikationen mit Infarkten oder Organversagen kann eine Austauschtransfusion erforderlich sein.
Entscheidend für die Prognose der Patienten ist die Vermeidung oder Aktubehandlung von Komplikationen. Rechtzeitige Antibiotikatherapie bei Infekten und prophylaktische Impfungen sind Teil des Behandlungskonzeptes. Die Leitlinie en werden regelmäßig ergänzt und sind über das Internet bei großen Zentren in USA und Großbritannien einsehbar (z.B. http://www.nhlbi.nih.gov/health/prof/blood/sickle/sc_mngt.pdf, http://www.sicklecelldisease.org )
Einzig potentiell kurative (auf Heilung ausgerichtete) Behandlung ist bislang die Knochenmark- oder Blutstammzelltransplantation. Wegen der damit verbundenen Gefahren, wird sie lediglich bei schweren Formen vor eingesetzt.
Membrandefekte
Die häufigsten Defekte in der Erythrozytenmembran führen zur Kugelzellanämie (hereditäre Sphärozytose). Sie ist die häufigste angeborene Form einer hämolytischen Anämie in Mitteleuropa. Die Diagnose erfolgt teilweise erst im Erwachsenenalter Anhand der typischen Konstellation mit Hämolyse (oft kompensiert), Milzvergrößerung und Nachweis von Gallensteinen. Bestätigt wird die Diagnose mittels Nachweis von Kugelzellen im Blutausstrich, Bestimmung der osmotischen Resistenz und positivem Eosin-Malein-Test (EMA-Test). Eine Behandlung Entfernung der Milz (Splenektomie) normalisiert fast immer das Blutbild, ist aber nur bei schwerer Hämolyse erforderlich. Daneben gibt es eine Reihe anderer Membranstörungen wie die Elliptozytose und die Stomatozytose, die durch den Nachweis charakteristischer Veränderungen im Blutausstrich und /oder durch spezielle Teste diagnostiziert werden können.
Hämolytische Anämien durch Enzymdefekte
Angeborene Enzymvarianten mit in der Folge verminderter Enzymaktivität führen zu einem vorzeitigen Abbau der Erythrozyten. Am häufigsten ist der Mangel an Pyruvatkinase und Glukose-6-Phosphatdehydrogenase. Seltener sind Defekte der Hexokinase, Triosephosphatisomerase, Diphosphoglyceratmutase sowie der Glutathionreduktase.
Instabile Hämoglobine
Unter diese Bezeichnung fällt eine Gruppe von Erkrankungen, die sich durch Änderung der Aminosäuresequenz des Hämoglobinmoleküls auszeichnen und zu einer deutlich verkürzten Überlebenszeit der Erythrozyten führen. Die Veränderungen werden nach den Orten der Erstbeschreibung benannt (z.B. Hb-Köln). Die Diagnose erfolgt durch die Hämoglobinanalyse. Allen gemeinsam ist eine Hämolyse mit Milzvergrößerung. Auslöser der Hämolyse sind oft Medikamente. Bei schwerer hämolytischer Anämie kann die Milzentfernung notwendig werden.
Sideroblastische (sideroachrestische) Anämien
Die sideroblastischen Anämien sind eine Gruppe von Erkrankungen die sich durch eine ausgesprochene Ineffektivität der Hämopoese auszeichnen. Zugrunde liegen sowohl genetische Ursachen (z.B. ALAS2-Mutation bei der hereditären sideroblastischen Anämie) als auch erworbene Faktoren (Medikamente, Bleivergiftung).
Kongenital dyserythropoetische Anämien
Sehr selten sind die (angeborenen) kongenital dyserythropoetischen Anämien (CDA), die anhand von Veränderungen im Knochenmark und Blut diagnostiziert werden können. Unter dem Begriff CDA werden mehrere Erkrankungen mit den unterschiedlichen Schweregraden zusammengefasst. Derzeit sind im deutschsprachigen Raum weniger als 150 Patienten bekannt. Gekennzeichnet ist die Erkrankung durch eine ineffektive Blutbildung mit führender Anämie. Der größte Teil der Patienten hat eine normale Lebenserwartung, ist aber im Laufe seines Lebens durch Komplikationen (Gallensteinbildung, Einsenüberladung, aplastische Krisen) gefährdet.
Therapie der angeborenen Anämien
Über die Behandlung muss je nach Krankheitsgruppe und Schweregrad individuell entschieden werden. Dies gilt unter anderem für die Indikationsstellung zur Milzentfernung (Splenektomie), für die der Rat des Spezialisten erforderlich ist. Bei Patienten mit Eisenüberladung ist ein frühzeitiger Beginn einer Eisenentzugsbehandlung (Eisenchelation) notwendig.
Red Cell Center Ulm

Das Red Cell Center Ulm (RCCU) ist ein Zusammenschluss von Ärzten der Kinder- und Jugendmedizin, Transfusionsmedizin und Inneren Medizin/Hämatologie. Ziel ist die integrative Versorgung von Patienten mit angeborenen und erworbenen Anämien insbesondere die Sicherstellung einer nahtlosen Betreuung beim Übergang von der Pädiatrie zur Inneren Medizin.