Genomically Instable Pancreatic Cancer
Pancreatic ductal adenocarcinoma (PDAC) is the most commonly diagnosed type of pancreatic cancer. It is a devastating disease carrying a dismal clinical prognosis, rapidly progressing and highly therapeutic resistant, as reflected by its very poor five-year overall survival that just recently reached 10%. With an incidence increasing by 0.5-1.0% per year, it is projected to become the second-leading cause of cancer-related deaths by 2030 in the Western World, surpassing then colorectal and breast cancers. Human PDAC is a genetically complex and remarkably heterogeneous disease, displaying a multitude of genomic alterations and dysregulation of major operative signaling networks, leading to multiple biological effects, and supporting tumor progression and dissemination.
Complex rearrangement patterns and mitotic errors are hallmarks of PDAC. Among DNA lesions, DNA double-strand breaks (DSB) bear the greatest risk of provoking genomic instability. Two major repair pathways dominate DSB repair for safeguarding the genome integrity: non-homologous end joining and homologous recombination (HR). Alterations in HR genes (e.g. ATM, BRCA1/2, PALB2) affect approximately 15% of all PDAC cases. They result in an inability to repair DSBs that enhances both genomic instability and mutational rate, which eventually drives tumor evolution and progression, but also sensitizes cells to DNA-damaging chemotherapies. While the loss of ATM accelerates cancer formation and favors epithelial-to-mesenchymal transition and cancer aggressiveness (Russell, 2015; Perkhofer, 2017), the subsequent HR deficiency (HRD) constitutes a vulnerability that can be therapeutically exploited to eradicate HRD tumor cells (Perkhofer, 2017; Roger, 2020; Gout, 2021).


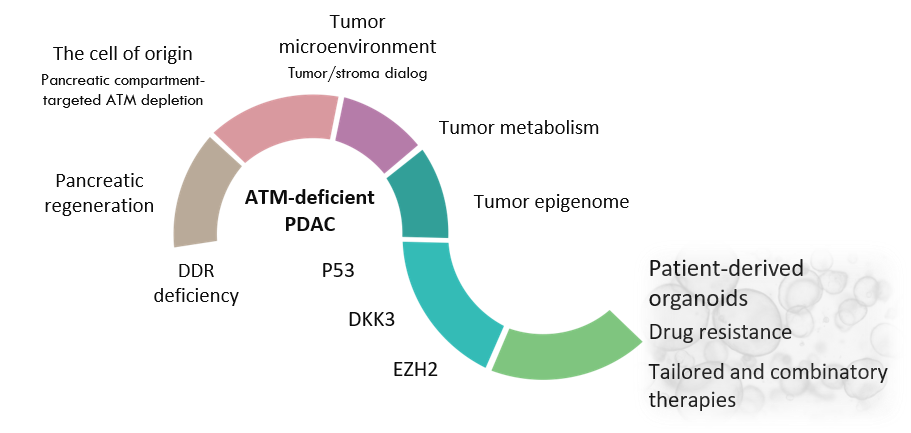
Employing state-of-the-art mouse and human models (genetically engineered mouse models, preclinical mouse models as syngeneic orthotopic mouse models and patient-derived xenografts, patient-derived tumor organoid living biobank), our translational approach aims to further understand the exceptional biology and pathology of HRD PDAC and dissect the molecular mechanisms underpinning its initiation and progression, to identify novel actionable vulnerabilities and thereby, design innovative targeted therapeutic strategies.
Some of the research projects currently being developed in the group are the followings:
Research Topics
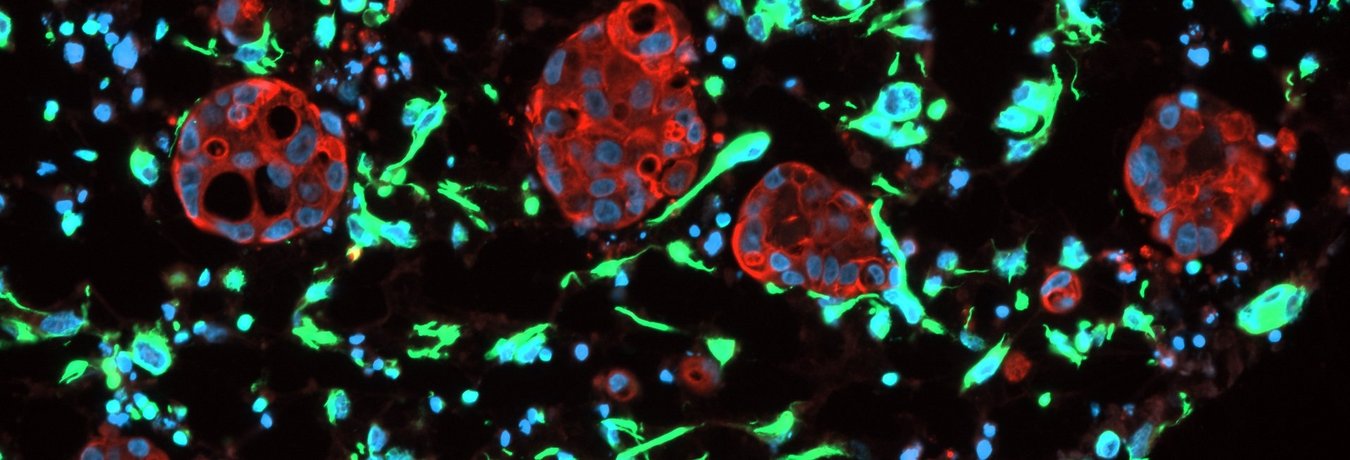
It has become evident that pancreatic cancer initiation and progression also relies on tumor microenvironment (TME) including activated fibroblasts, infiltrating immune cells, neurons, and extracellular matrix components. These TME compartments and the neoplastic cells interact with each other providing both promotive and suppressive tumor signals. Such tumor–stroma dialog can occur directly via cell–cell interaction or via multiple secreted molecules. This secretome, which derives not only from tumor cells but also from cancer-associated stromal cells, is an important source of key regulators of the tumorigenic process.
Our research group focuses on deciphering the molecular cascades and gene regulatory networks underlying specific tumor–stroma crosstalks and TME shaping in HRD pancreatic cancers as well as their subsequent impact on the progression and dissemination of these particularly aggressive subtypes. Our multidisciplinary approach combining the use of genetically engineered mouse models of pancreatic cancer, ex vivo and in vitro 3D co-culture systems involving patient-derived organoids, and omics and single-cell sequencing approaches will chart a comprehensive molecular and cellular picture of the tumor–stroma signaling in HRD PDACs to therefore support the identification of novel promising therapeutic targets.
The massive PDAC desmoplastic reaction results in harsh tumor microenvironments characterized by severe
hypoxia, nutrient deprivation, and oxidative stress. To sustain their survival and unlimited proliferation in such nutrient-depleted environments, pancreatic cancer cells adapt by rewiring many metabolic pathways.
ATM, initially described to regulate DNA damage repair and cell cycle arrest, was further described to modulate cell metabolic functions through its non-canonical signaling pathways. We are currently exploring nutrient stress adaptation and metabolic reprogramming in ATM-deficient malignant cells and how it fosters cancer progression and metastatic dissemination, with the overarching goal to reveal novel workable metabolic vulnerabilities to treat HRD patients.
Genomic alterations do not fully explain the entire pancreatic cancer heterogeneity, highlighting other mechanistic drivers like epigenetics. In a highly dynamic manner, an intricate network of transcription factors and epigenetic regulatory proteins temporally and spatially controls cell transcriptomic states contributing to PDAC heterogeneity. Given the reversibility of epigenetic processes, key epigenetic drivers have recently emerged as promising novel therapeutic targets in pancreatic cancer.
The chromatin regulatory protein and histone methyltransferase enhancer of zeste homolog 2 (EZH2) plays a pivotal role in ataxia telangiectasia where its depletion rescues ATM deficiency-mediated neurodegeneration, underpinning the impact of epigenetic dysregulation in ATM-deficient context. In PDAC, EZH2 expression and hyperactivity fosters aggressiveness and chemoresistance. Together with the team of our long-lasting collaborator Prof. Dr. Elisabeth Hessmann (University Medical Center of Göttingen), we thus particularly investigate the role of EZH2 in a homologous recombination-deficient PDAC subtype lacking ATM and the possible therapeutic implications of the EZH2–ATM axis.
Distinct cell types comprise the pancreatic niche and its reprogramming is a strong pathogenic trigger during inflammatory pancreatic disease and pancreatic cancer.
Accumulating evidences suggest that the cancer cell-of-origin within the pancreas determines distinct PDAC biology and subtypes. As a consequence, the cellular context of investigating the potential of an oncogene or tumor suppressor is highly critical to determine its function during pancreatic cancer initiation, progression, and maintenance. Using spatiotemporal deletion strategies in vivo and in vitro, current research in our group focuses on identifying the most permissive cell type to become dysplastic upon ATM loss and on assessing to what extent does such difference in the cell type of origin affects PDAC biology, to better steer tailored therapies for ATM-mutated patients.