AG Prof. Dr. Schirmbeck
Many vaccines are designed to prime multi-specific CD8 T cell responses, because the cellular immune system mediates protective immunity to many intracellular pathogens and tumours. We have established novel DNA- protein- and peptide-based vaccination strategies to induce CD8 T cell responses against viral, tumor and ´self´ antigens in different mouse models. In particular, we designed antigens that target endogenous “helper” molecules (endogenous adjuvant) and thereby facilitate priming of cellular and humoral immune responses under difficult conditions.

Preclinical mouse tumor models:
CD8 T cells play a crucial role for the control of many human and murine tumors. In an optimal scenario, the immune system specifically detects and eliminates aberrant tumor progenitor cells before they develop into clinically manifested tumors. This implied that tumor cells per se are immunogenic and can present tumor-associated antigens (TAAs) to the immune system in a manner that allows priming of effector CD8 T-cell responses. However, many tumors developed mechanisms to escape from recognition and elimination by CD8 T cells. Downregulation or loss of molecules involved in MHC-I processing/presentation was evident in many primary human and murine cancer cells, thereby limiting their presentation-competence to CD8 T cells. Furthermore, PD-L1 expression on tumor cells could function as a molecular shield and directly silence effector T-cell responses through co-inhibitory PD-L1/PD-1 interactions. Tumors like pancreatic ductal adenocarcinoma (PDAC) also generate an immune-suppressive microenvironment, composed among others of PD-L1 expressing myeloid-derived suppressor cells and/or regulatory CD4+ Foxp3+ Tregs that may enhance the down-regulation of tumor-specific CD8 T-cell responses and/or affect the limited responsiveness of murine and human PDAC to antibody-mediated PD-L1/PD-1 check point inhibition.
We generate tumor antigen-expressing vectors, immunize PD-L1/PD-1 competent/-deficient mice and determine priming of tumor-specific CD8 T cell responses. Furthermore, we test the potential of tumor cells per se to prime tumor-protective CD8 T cells. In this cell-based immunization strategy, we further test different manipulations of the tumor cells (e.g., deletion of co-inhibitory PD-L1 signaling).

Induction and suppression of autoreactive CD8 T cells directed against beta cell antigens.
DNA vaccines against autoimmune type 1 diabetes (T1D) contain a non-predictable risk to induce autoreactive T-cell responses rather than a protective immunity. Little is known if (and how) antigen expression/processing requirements favor the induction of autoreactive or protective immune responses by DNA immunization. We analyse whether structural properties of preproinsulin (ppins) variants and/or subcellular targeting of ppins designer antigens influence the priming of effector CD8+ T-cell responses by DNA immunization. Primarily, we use H-2b RIP-B7.1 tg mice, expressing the co-stimulator molecule B7.1 in beta cells, to identify antigens that induce or do not induce autoreactive ppins-specific (Kb/A12-21- and/or Kb/B22-29) CD8+ T-cell responses. Female NOD mice, expressing the diabetes-susceptible H-2g7 haplotype, were used to test ppins variants for their potential to suppress spontaneous diabetes development. We show that ppins antigens excluded from expression in the Endoplasmic Reticulum (ER) did neither induce CD8+ T cells nor autoimmune diabetes in RIP-B7.1 tg mice, but efficiently suppressed spontaneous diabetes development in NOD mice as well as ppins-induced CD8+ T-cell-mediated autoimmune diabetes in PD-L1-/- mice. The induction of a ppins-specific therapeutic immunity in mice has practical implications for the design of immune therapies against T1D in individuals expressing different MHC I and II molecules.
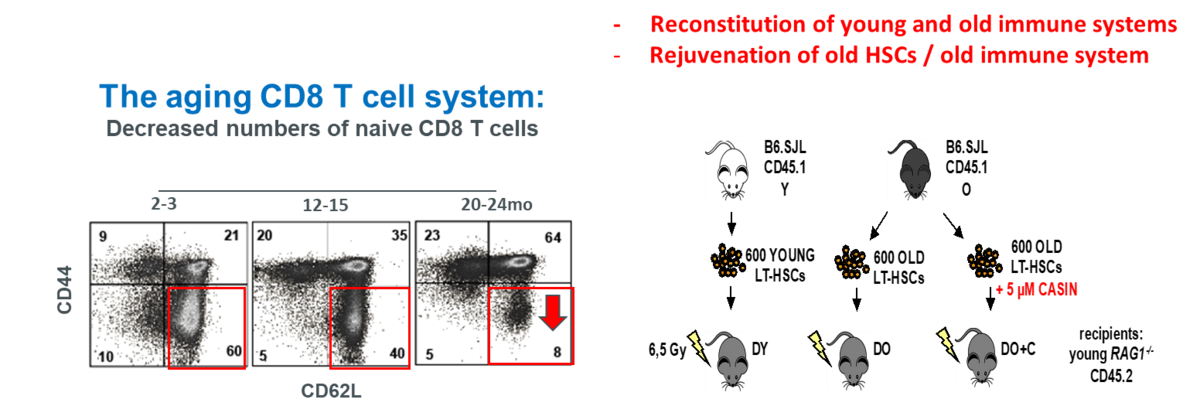
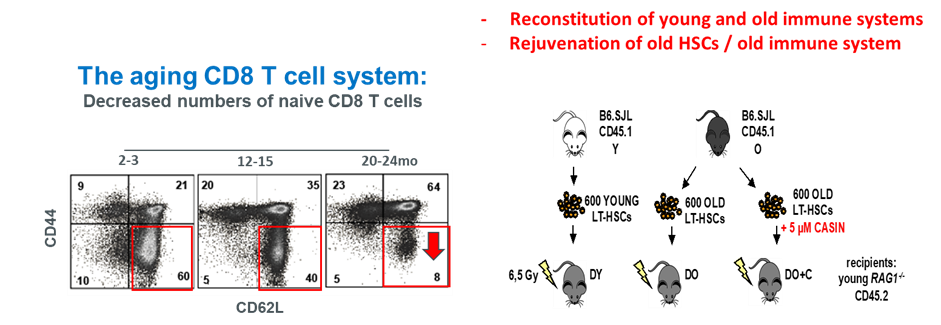
Aged Murine Hematopoietic Stem Cells Drive Aging-Associated Immune Remodeling
Aging-associated remodeling of the immune system impairs its functional integrity and contributes to increased morbidity and mortality in the elderly. Aging of hematopoietic stem cells (HSCs), from which all cells of the adaptive immune system ultimately originate, play a crucial role in the remodeling of the aged immune system. We recently reported that aging of HSCs is, in part, driven by elevated activity of the small RhoGTPase Cdc42 and that aged HSCs can be rejuvenated in vitro by inhibition of the elevated Cdc42 activity in aged HSCs with the pharmacological compound CASIN.
To study the quality of immune systems stemming selectively from young or aged HSCs, we established a HSC-transplantation model in T- and B-cell deficient young RAG1-/- hosts. We show that both phenotypic and functional changes in the immune system upon aging are primarily a consequence of changes in the function of HSCs upon aging and to a large extent independent of the thymus, as young and aged HSCs reconstituted distinct T- and B-cell subsets in RAG1-/- hosts that mirrored young and aged immune systems. Importantly, aged HSCs treated with CASIN re-established an immune system similar to the one of young animals and thus capable of mounting a strong immune response to vaccination. Our studies further imply that epigenetic signatures already imprinted in aged HSCs determine the transcriptional profile and function of HSC-derived T- and B-cells.
Rejuvenation of the aged immune system by depletion of Asialo GM1-expressing memory CD8+ T cells
A prominent hallmark of senescent immune remodeling is a significant decrease in naïve (CD44lo CD62Lhi) CD8+ T cells, ranging from 80% in young (2-3 months) to < 10% in old (> 20 months) mice, followed by a concomitant increase in aging-associated memory (CD44hi CD62Lhi)CD8+ T cells. As a possible consequence, vaccines usually prime antigen-specific effector CD8+ T cells in young but not (or inefficiently) in old mice. Little is known if (and how) the accumulation of memory CD8+ T cells influences naïve T cell functions in the old.
We identified a novel cell surface marker (the ganglioside asialo GM1; GA1) specifically presented with memory CD8+ T cells that correspond to about 15% or 80% of total CD8+ T cells in unimmunized young and old mice, respectively. GA1 is not (or barely) expressed on naïve CD8+ T cells, but rapidly induced on both, young and old CD8+ T cells upon activation (e.g., by stimulation of naïve CD8+ T cells with anti CD3/CD28 antibodies in vitro). Injection of anti GA1 antibody into vaccinated mice efficiently suppressed effector CD8+ T-cell responses. In contrast, depletion of memory CD8+ T cells with anti GA1 antibody prior to vaccination enhanced the priming of HBV-Core (Kb/C93-100)-specific effector CD8+ T cells in old but not in young mice. Injection of anti GA1 antibody into unimmunized old mice resulted in an efficient and long-lasting depletion of memory CD8+ T cells, generating a youthful CD8+ T cell compartment presented primarily with naïve cells that re-established a youthful antigen-specific CD8+ T cell response to DNA vaccination. Our studies imply that memory CD8+ T cells in unimmunized old (but not in young) mice play a crucial role in the down-regulation of de novo primed effector CD8+ T cells by vaccination.
Priming of COVID-19 specific immune responses in aging mice.
A progressive decline in the ability to mount protective immune responses is observed with advancing age. Clinical trials revealed deficiencies in the elderly that limit priming of protective, specific immunity by vaccines. Though limited in some respects, the mouse (as an immunologically well defined preclinical model) can provide important insight into age-associated changes in the T and B cell system and the evaluation of potent vaccine strategies.
We develop novel vaccination strategies that efficiently induce antigen-specific CD8 T cell- and humoral immune responses in old mice. Vaccine-relevant antigens (e.g., COVID-19 antigens) are designed and delivered either as DNA-based vaccines or as recombinant protein antigens into aging mice. In conventional approaches of protein delivery, we use various adjuvant formulations that stimulate different arms of the innate immune system (e.g., RNA/TLR-7, CpG-containing oligonucleotides/TLR-9) and/or that modulate uptake and longevity of the antigens (e.g., ISCOM-derivates; alum). However, we also use novel strategies in which vaccine antigens per se target cellular adjuvant-like “helper molecules” (cellular RNA or cellular stress proteins) that efficiently enhance priming of CD8 T cell- and antibody responses in both, young and aged mice.




