Myeloproliferative Neoplasie (MPN)
Ansprechpartner
Spezialsprechstunde MPN
Anmeldung zur Spezialsprechstunde unter Infos für Ärzte
Labor
Therapie und Forschung in der Klinik für Innere Medizin III
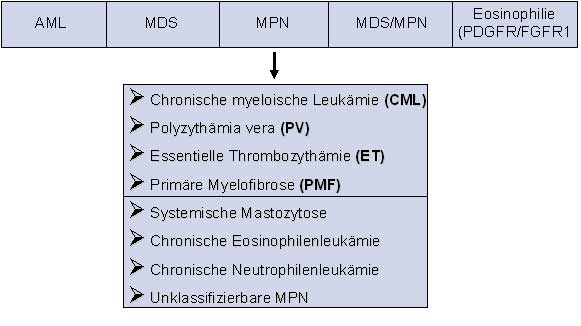
In der Klinik für Innere Medizin III werden alle Formen myeloproliferativer Neoplasien (MPN) behandelt. Zu den häufigsten MPN zählen die chronische myeloische Leukämie (CML), die essentielle Thrombozythämie (ET), die Polyzythämia vera (PV) und die primäre Myelofibrose (PMF) (Abb. 1). Seltenere MPN sind z.B. die systemische Mastozytose (SM) oder die chronische Eosinophilenleukämie (CEL). Ein besonderer Schwerpunkt liegt auf der Durchführung und Beteiligung an klinischen Studien, mit dem Ziel die derzeitigen Therapiestandards zu verbessern und den Patienten neu entwickelte Medikamente vor deren Zulassung zur Verfügung stellen zu können. Aufgrund des verbesserten Verständnisses der Biologie der Erkrankungen wird bei MPN zunehmend eine zielgerichtete Therapiestragie verfolgt. Das Zentrum Ulm beteiligt sich intensiv an den klinischen Studien der deutschen CML-Studiengruppe (CMLSG). Um auch die Therapie von ET, PV und PMF weiter zu verbessern, wurde 2008 die MPN-Studiengruppe (GSG-MPN) gegründet. Im Jahr 2009 konnte die erste Studie der GSG-MPN aktiviert werden. Ulm ist Mitbegründer der Studiengruppe und nationales Referenzlabor. Die Betreuung von Patienten mit MPN findet überwiegend in der Spezial-Ambulanz statt.
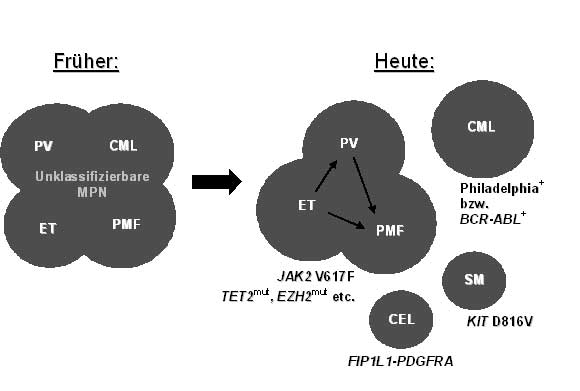
Ein wichtiger Schwerpunkt der Klinik für Innere Medizin III liegt in der Erforschung der genetischen Grundlagen dieser Erkrankungen. Hierzu werden von der Erkrankung betroffene Knochenmark und/oder Blutzellen von Patienten auf das Vorliegen von genetischen Veränderungen untersucht. In den letzten Jahren konnten große Fortschritte in der genetischen Charakterisierung der MPN gemacht werden. Mittlerweile sind eine Reihe von genetischen Veränderungen bei MPN-Patienten bekannt, die eine genauere Abgrenzung der Erkrankungen von einander erlauben. Auf diese Weise kann nicht mehr nur die CML aufgrund des Vorliegens des sog. Philadelphia-Chromosoms von den anderen MPN unterschieden werden, sondern es ist auch möglich geworden, die Philadelphia-negativen MPN auf Grundlage der vorliegenden genetischen Veränderungen einfacher zu diagnostizieren und besser zu klassifizieren. Beispielhaft kann hier die Identifizierung von Mutationen in den Genen JAK2, TET2 und EZH2 genannt werden (Abb. 2). Diese genetische Klassifizierung wird in den nächsten Jahren auch die Therapie der MPN wesentlich beeinflussen. Darüber hinaus ermöglicht die stetige Weiterentwicklung neuer Technologien die Erforschung bislang unbekannter genetischer Veränderungen.
Sowohl die klinischen Studien als auch die Grundlagenforschung werden in enger Zusammenarbeit mit deutschen oder internationalen Studiengruppen durchgeführt. Ziel ist es, dass die Ergebnisse dieser Forschung dazu beitragen, in Zukunft effektivere Therapien zu finden, die auf die besonderen genetischen Merkmale der einzelnen Patienten abgestimmt sind.
Klinische Studien
Weitere Informationen zu den aktiven Studien erhalten Sie unter:
Beschreibung der Erkrankung
Basisinformation
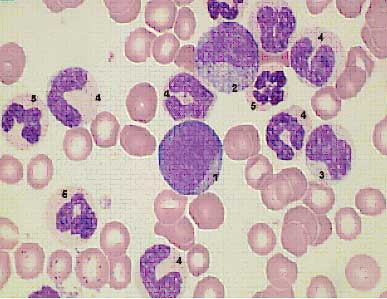
MPN sind bösartige Knochenmarkerkrankungen (Ort der Blutbildung) mit langsamem Verlauf. Das klinische Spektrum der Erkrankungen ist vielfältig. Die CML ist typischerweise durch eine starke Vermehrung weißer Blutkörperchen (Leukozyten) in unterschiedlichen Ausreifungsformen gekennzeichnet (Abb. 3). Unbehandelt geht die schleichende Erkrankung („chronische Phase“) nach einigen Jahren in eine bedrohlichere und schneller verlaufende Form über („Akzelerationsphase“) und verläuft zuletzt wie eine akute Leukämie („Blastenkrise“). In über 90% der Fälle liegt der Erkrankung eine definierte genetische Veränderung zugrunde, das sog. Philadelphia-Chromosom. Dieses entsteht durch Austausch von Erbmaterial zwischen den Chromosomen 9 und 22. Mittlerweile hat man auf der Grundlage diesr genetichen Veränderung Substanzen entwickelt, die sehr spezifisch und zielgerichtet wirken. Hierbei handelt es sich um die sog. Tyrosinkinasinhibitoren (TKI). In späteren bzw. fortgeschritteneren Erkrankungsstadien treten neben dem Philadelphia-Chromosom noch weitere genetische Veränderungen auf. Dies belegt, dass die Erkrankung auch auf biologischer Ebene fortschreitet.
Bei der ET steht die Vermehrung von Blutplättchen (Thrombozyten) im Vordergrund der Erkrankung. Bei der PV ist eine Erhöhung der Anzahl roter Blutkörperchen (Erythrozyten) typisch, allerdings können zusätzlich auch Thrombozyten und Leukozyten vermehrt sein. In der Frühphase der PMF ist wie bei der ET die Anzahl der Thrombozyten im Blut erhöht, im weiteren Verlauf kommt es jedoch zu einem zunehmenden bindegewebigen Umbau des Knochenmarks mit der Folge von Zellarmut. Neben diesen drei klassischen Formen der Philadelphia-negativen MPN gibt es auch fließende Übergänge zwischen der ET, der PV und der PMF. Die genetischen Ursachen von ET, PV und PMF sind im Gegensatz zur CML komplex. Allerdings ist es in den letzten Jahren gelungen, verschiedene Genmutationen zu identifizieren, die die Krankheitsentstehung von ET, PV und PMF zumindest teilweise erklären. Z.B. findet man bei fast allen Patienten mit PV und bei etwa 50% der Patienten mit ET und PMF die Mutation V617F im Tyrosinkinase-Gen JAK2. Derzeit sind zahlreiche Forschungsprojekte aktiviert, die zum Ziel haben, sich diese Erkenntnisse wie bei der CML therapeutisch zu Nutze zu machen. Hierzu sollen zielgerichtete Medikamente entwickelt werden, die die Vermehrung der erkrankten Zellen hemmen. In Deutschland erkranken ca. 1-2 Erwachsene pro 100.000 Einwohner pro Jahr an einer MPN. Das Risiko nimmt mit steigendem Lebensalter zu. Die Erkrankungen können aber in allen Altersgruppen auftreten.
Krankheitszeichen
Symptome und Befunde
Da Blutuntersuchungen heutzutage regelmäßig durchgeführt werden, können MPN bei einem Großteil der Patienten früh diagnostiziert werden. Viele Patienten haben von der Erkrankung zum Zeitpunkt der Diagnose noch nichts bemerkt. Neben den oben geschilderten Blutbildveränderungen sind Müdigkeit, Leistungsminderung und Nachtschweiß mögliche Symptome. Bei ET und PV sind Durchblutungsstörungen in kleinen Gefäßen (Kribbeln in Händen und Füßen) bis hin zu venösen oder arteriellen Gefäßverschlüssen (Thrombosen, Thrombembolien) möglich. Insbesondere in fortgeschritteneren Stadien ist bei MPN regelmäßig eine Milzvergrößerung vorhanden, die zu einem Druckgefühl im linken Oberbauch führen kann.
Diagnosesicherung
Die Diagnosestellung erfolgt nach WHO-Kriterien durch Untersuchung von peripherem Venenblut und Knochenmark. Hieraus werden neben den mikroskopischen Befunden auch die genetischen Charakteristika der Erkrankung untersucht.
Klassifikation
Die Klassifikation erfolgt anhand der oben dargestellten WHO-Einteilung. Die CML wird anhand von Werten aus Blut- und Knochenmark in die unterschiedlichen Erkrankungsphasen eingteilt (s.o.). Bei ET, PV und PMF erfolgt aufgrund der Blutwerte und der Erkrankungsgeschichte des Patienten die Einteilung in eine Niedrig-, Mittel-, oder Hochrisiko-Gruppe. Diese Unterteilung hat wesentlichen Einfluss auf die jeweilig empfohlene Therapie.
Therapeutische Möglichkeiten
Durch den oben erwähnten Einsatz von TKI hat sich die Therapie der MPN in den letzten Jahren zunehmend geändert. Insbesondere bei der CML hat das Prinzip der molekular zielgerichteten Therapie die Behandlung revolutioniert. Die Wirkung von TKI beruht auf der gezielten Behandlung der Veränderungen in den erkrankten Zellen, die durch das Philadelphia-Chromosom entstehen. TKI werden in Tablettenform eingenommen. Am häufigsten wird Imatinib (Handelsname Glivec®) eingesetzt. Nebenwirkungen wie Übelkeit, Muskelkrämpfe und Flüssigkeitsansammlungen im Gewebe (Ödeme) treten bei einem Teil der Patienten auf. Die regelmäßige, tägliche Einnahme des Medikaments ist dauerhaft notwendig, da die CML nur so effektiv zurückgedrängt und das Fortschreiten der Erkrankung verhindert werden können. Wichtig ist außerdem, dass die Therapie unmittelbar nach Diagnosestellung begonnen wird, da die CML in weiter fortgeschrittenen Stadien schwerer behandelbar ist als in der frühen Phase. In den meisten Fällen normalisieren sich schon nach wenigen Wochen Therapie die weißen Blutkörperchen. Darüber hinaus wird der Behandlungserfolg durch regelmäßige Blutuntersuchungen mittels moderner Labormethoden exakt bestimmt. Bei einigen Patienten spricht die Behandlung so gut an, dass nach Monaten der Therapie kaum noch oder keine Krankheitsaktivität mehr nachweisbar ist. Trotzdem liegt nach derzeitigen Erkenntnissen keine Heilung der CML vor, da die Erkrankung nach Absetzen des Medikaments rasch wiederkehrt und dann unter Umständen schlechter behandelbar ist. Problematisch ist, dass es unter Therapie mit TKI zur Resistenz der erkrankten Zellen gegenüber dem Medikament kommen kann. Möglicherweise wirken neuere TKI, die momentan klinisch erprobt werden, noch nachhaltiger auf die erkrankten Zellen bei gleichzeitig besserer Verträglichkeit. Nilotinib (Tasigna®) und Dasatinib (Sprycel®) sind zwei vielversprechende TKI der neueren Generation, die in Studien untersucht werden. Ulm ist teilnehmendes Zentrum einer CMLSG-gestützten Studie, die den Einsatz von Nilotinib bei neu diagnostizierten CML-Patienten ermöglicht. Durch den Einsatz der TKI sind intensive und nebenwirkungsreichere Therapieformen wie Chemotherapie und Blutstammzelltransplantation deutlich in den Hintergrund gerückt. Auf diese kann jedoch bei TKI-Unverträglichkeit oder -Versagen zurückgegriffen werden.
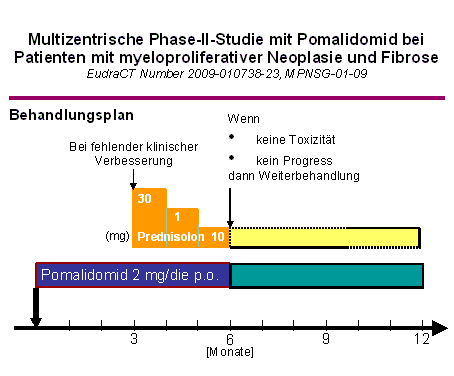
Manche Formen seltener MPN, wie z.B. die CEL mit Nachweis der Genfusion FIP1L1-PDGFRA, sprechen ebenfalls auf eine Therapie mit Imatinib an. Dies gilt jedoch nicht für Patienten mit ET, PV und PMF, da die Mutation V617F im JAK2-Gen nicht Imatinib-empfindlich ist. Unterschiedliche JAK2-Inhibitoren sind zur Zeit in klinischer Erprobung. Erste Studienphasen zu den Substanzen sind abgeschlossen. Zum Ende des Jahres 2010 werden erneute Studienaktivitäten erwartet, die die Behandlung von MPN-Patienten mit einem JAK2-Inhibitor ermöglichen. Neben den JAK2-Inhibitoren sind derzeit weitere Substanzen wie z.B. die IMiDe, insbesondere bei MF-Patienten in Erprobung. MF-Patienten können, wenn Sie die Einschlusskriterien erfüllen, derzeit in die MPNSG-01-09 Studie eingeschlossen werden (Abb. 4). Hierbei handelt es sich um eine einarmige Studie mit Pomalidomid, die im Dezember 2009 aktiviert wurde. Pomalidomid gehört zu der Gruppe immunmodulatorischer Medikamente. Hintergrund für den Einsatz von IMiDen sind Daten, die eine Verbesserung der Blutbildung bei MF-Patienten aufzeigen. Die als Tablette verfügbare Substanz scheint insbesondere wirksam bei der Behandlung von Blutarmut und erniedrigten Thrombozytenzahlen zu sein.
Außerhalb von Studien richtet sich die Behandlung von ET-, PV- und PMF-Patienten nach etablierten Risikomerkmalen, die sich aus der Krankengeschichte des Patienten, dem Lebensalter und den Blutbildwerten ableiten. Durch die Erhebung dieser Parameter kann der Verlauf der Erkrankung individuell besser eingeschätzt werden. Bei ET-Patienten mit niedrigem Risiko kann die alleinige Beobachtung der Blutbildwerte ausreichend sein. Bei PV-Patienten werden in der Regel Aderlässe zur Senkung der Anzahl von Erythrozyten durchgeführt und Acetylsalicylsäure in niedriger Dosierung (100 mg täglich) verabreicht. Im Falle eines erhöhten Risikos für thrombembolische Komplikationen können zusätzlich Medikamente eingesetzt werden, die die Thrombozyten und/oder Erythrozyten im Blut reduzieren. Beispielhaft können hier Hydroxyurea und Interferon-alpha genannt werden. Bei PMF-Patienten kann anhand der Risikostratifikation insbesondere die Prognose der Erkrankung abgeschätzt werden. Bei Patienten mit ungünstiger Konstellation, bei denen ein rasches Voranschreiten der Erkrankung wahrscheinlich ist, kann dann z.B. die Empfehlung zur Verabreichung einer intensiveren Chemotherapie und nachfolgender Durchführung einer Blutstammzelltransplantation ausgesprochen werden. Aufgrund der starken Nebenwirkungen muss hier eine besonders sorgfältige Nutzen-Risiko-Abwägung erfolgen.
Förderung der MPN-Forschung an der Universitätsklinik Ulm
• Else Kröner-Fresenius-Stiftung