Akute Leukämien
Ansprechpartner

Prof. Dr. med. Hartmut Döhner
Ärztlicher Direktor der Klinik für Innere Medizin III (Hämatologie, Onkologie, Palliativmedizin, Rheumatologie und Infektionskrankheiten)
Schwerpunkte
Stv. Direktor, Comprehensive Cancer Center Ulm (CCCU)
Mitglied des Direktorats des Nationalen Centrums für Tumorerkrankungen SüdWest (NCT SüdWest)
Leiter der Deutsch-Österreichischen AML Studiengruppe (AMLSG)


Spezialsprechstunde AML
Anmeldung zur Spezialsprechstunde unter Infos für Ärzte
Ansprechpartner Studienzentrale
Diagnostik, Therapie und Forschung in der Klinik für Innere Medizin III
Klinik für Innere Medizin III – eine weltweit renommierte Institution
Die Klinik für Innere Medizin III ist national und international eine der renommiertesten Einrichtungen zur Erforschung, Diagnostik und Behandlung der akuten myeloischen Leukämie (AML). Die Klinik ist Hauptsitz der Deutsch-Österreichischen AML Studiengruppe (AMLSG), mit derzeit ca. 80 Prüfzentren in Deutschland und Österreich eine der größten Studiengruppen weltweit. In der Klinik für Innere Medizin III werden jährlich etwa 150 Patienten mit neu diagnostizierter AML betreut.
Diagnostik
Die initiale Diagnostik erfolgt im Rahmen der AMLSG BiO (Biology and Outcome) Registerstudie (clinicaltrials.gov NCT01252485). In dieser Registerstudie geben die Patienten ihr Einverständnis zur genetischen Diagnostik, zur Dokumentation ihrer klinischen Daten und zu einem Biobanking, d.h. Zellproben werden für zukünftige Forschungsprojekte aufbewahrt. Die Diagnostik orientiert sich an den 2022 Empfehlungen des European LeukemiaNet (ELN) (Döhner at al. Blood 2022).
Die Diagnostik umfasst neben der morphologischen Begutachtung von Knochenmark- und Blutausstrichen, die immunphänotypische, die zytogenetische und molekulargenetische Diagnostik. Die morphologische und immunphänotypische Diagnostik erfolgt noch am Tag der Probenentnahme. Für die molekulargenetische Diagnostik haben wir seit vielen Jahren eine einzigartige diagnostische Plattform etabliert die es erlaubt, ein Panel von genetischen Markern (Gen-Fusionen, Gen-Mutationen) innerhalb von 24-48 Stunden zu analysieren und den Befund zu übermitteln. Die Etablierung dieses raschen molekularen Screenings wurde mit Einführung der ersten molekular zielgerichteten Therapien (v.a. der FLT3-Inhibitoren) notwendig. Das Ergebnis der zytogenetischen Analyse liegt innerhalb von 5-7 Tagen vor.
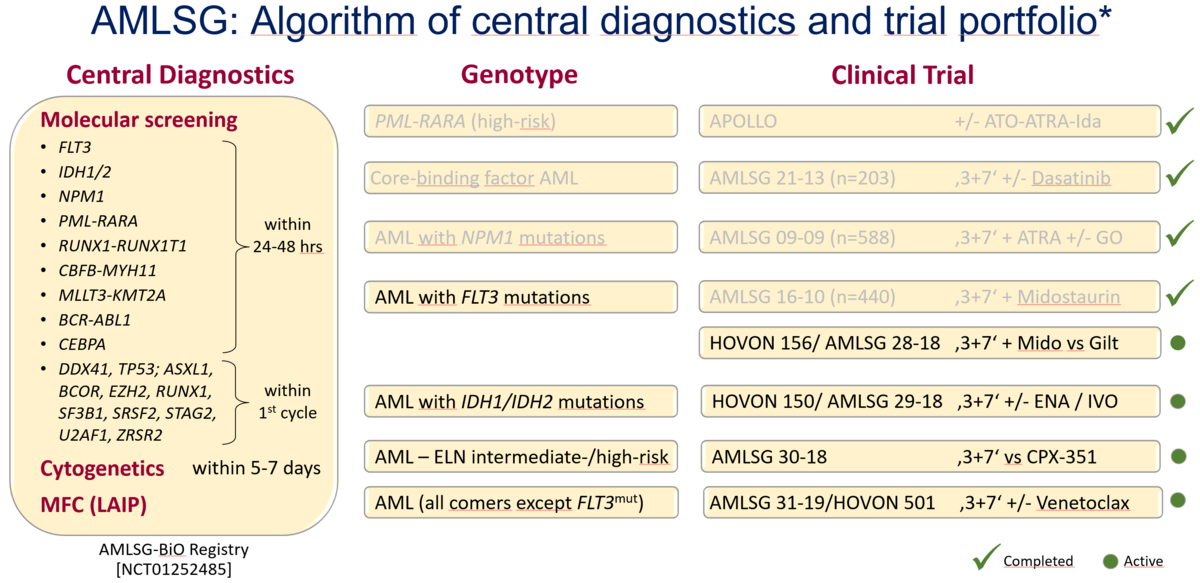
Genetische Risiko-Stratifikation
Die genetischen Marker sind nicht nur für die exakte Diagnose essentiell, sondern sie gehören auch zu den wichtigsten prognostischen Faktoren (siehe 2022 ELN Risikostratifikation). Beispielsweise dienen die genetischen Marker zur Entscheidungsfindung, ob einem Patienten eine allogene Blutstammzell-Transplantation empfohlen wird oder nicht. Darüber hinaus identifizieren die genetischen Marker in zunehmenden Maße Patienten für molekular zielgerichtete Therapien. Prominente Beispiele hierfür sind Mutationen in der Rezeptor-Tyrosinkinase FLT3 und den IDH Enzymen IDH1 und IDH2. Für diese Formen der AML sind mittlerweile erfolgreich Inhibitoren entwickelt und zugelassen worden.
Nachweis Minimaler Resterkrankung
Im Verlauf der Behandlung werden dann i.d.R. nach jedem Behandlungszyklus Knochenmark- und Blutproben auf minimale / messbare residuale Erkrankung (minimal or measurable residual disease [MRD]) untersucht. Grundsätzlich stehen für diese Analysen zwei Methoden zur Verfügung: molekulargenetische Assays (z.B. Nachweis von Gen-Fusionen oder Gen-Mutationen mittels hochsensitiver quantitativer PCR oder mittels Next-Generation Sequencing), und die Multi-Parameter Durchflusszytometrie.
Ausgewählte Forschungs-Projekte
Ein wichtiges Ziel unserer Forschungs-Projekte ist die Aufklärung der molekularen Pathogenese der Erkrankung. Hierzu gehören u.a. die Identifizierung und Charakterisierung von genetischen Markern, sowie die Ermittlung der prognostischen und prädiktiven Bedeutung dieser Marker. Zu diesem Zweck können wir auf die weltweit größte Biobank an primären Zell-Proben von AML-Patienten zurückgreifen. Es sind alle modernen Methoden der molekularen Genetik (z.B. konventionelle DNA-Sequenzierung, Next-Generation Sequenzierung, Polymerasen-Kettenreaktion [PCR], quantitative PCR, digitale PCR, Genexpressionsanalysen) sowie der Durchflusszytometrie etabliert.
Klinische Studien
Wenn immer möglich werden unsere Patienten im Rahmen von klinischen Therapiestudien betreut. Meist handelt es sich um innovative Therapiestudien, die wir unter unserer Federführung im Rahmen der AMLSG durchführen. Darüber hinaus sind wir aufgrund unseres Renommees und der großen Patientenzahl attraktiver Partner für die Pharmazeutische Industrie für Pharma-gesponserte Studien.
Weitere Informationen zu den aktiven Studien erhalten Sie unter:
Qualitätsmanagement
Unser Studienzentrum ist Teil des Zentrums für Klinische Studien unter dem Dach des Comprehensive Cancer Center Ulm (CCCU). Das Studienzentrum ist nach DIN EN ISO 9001:2008 zertifiziert und erfüllt die Forderungen des Kriterienkatalogs der Deutschen Gesellschaft für Hämatologie und Onkologie (DGHO).
Das Labor für Hämatologie, das Labor für Durchflusszytometrie sowie das Labor für Zytogenetische und Molekulargenetische Diagnostik sind nach DIN EN ISO 15189:2014 (Medizinische Laboratoriumsdiagnostik) und DIN EN ISO/IEC 17025:2005 (Medizinische Laboratoriumsuntersuchungen im Rahmen klinischer Studien) akkreditiert.
Beschreibung der Erkrankung
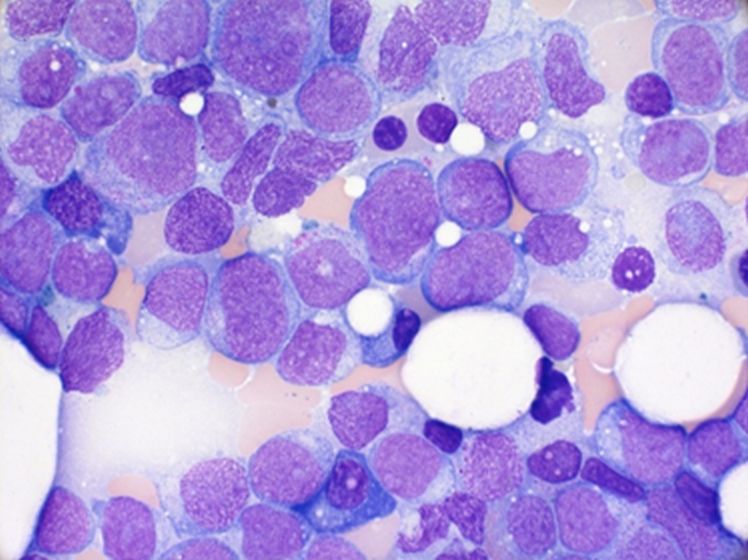
Die akute myeloische Leukämie (AML) ist eine bösartige Erkrankung des Knochenmarks. Wie andere Krebserkrankungen entsteht auch die Leukämie aus einer einzigen Zelle (klonale Erkrankung). Erworbene genetische Veränderungen (Mutationen) führen zu einer Reifungsstörung und ungehemmter Vermehrung funktionsuntüchtiger Blutzellen (sogenannte Blasten). Die Folgen sind eine Verdrängung der normalen blutbildenden Zellen (hämatopoetische Insuffizienz) und eine Ausschwemmung der Blasten in das Blut. Das Auftreten dieser Zellen im Blut führt zu einer weißlichen Verfärbung, weshalb Rudolf Virchow 1845 den Begriff Leukämie (= weißes Blut) einführte.
Häufigkeit, Erkrankungsalter und Risikofaktoren
Die Anzahl an AML-Neuerkrankungen wird derzeit mit etwa 2-4 Menschen je 100.000 Einwohner und Jahr angegeben und steigt kontinuierlich mit dem Lebensalter bis zu einem Erkrankungsgipfel von über 15 je 100.000 Einwohner und Jahr innerhalb der Personengruppe über 65 Jahre. Somit ist die AML eine Erkrankung des älteren Menschen, das mittlere Erkrankungsalter liegt bei ca. 70 Jahren.
Das Erbgut verändernde Substanzen sowie radioaktive Strahlung können das Risiko, an einer AML oder auch anderen Krebserkrankungen zu erkranken, erhöhen. Insbesondere nach vorangegangenen Strahlen- und Chemotherapien aufgrund einer zurückliegenden Krebserkrankung kommt es gehäuft zum Auftreten sogenannter Therapie-assoziierter myeloischer Erkrankungen. Des Weiteren kann sich aus einer bereits bestehenden Knochenmarkerkrankung, wie z.B. dem Myelodysplasie-Syndrom (MDS) oder Myeloproliferativen Neoplasien (MPN), eine sekundäre AML entwickeln.
Untersuchungen der letzten Jahre haben gezeigt, dass einige Patienten Keimbahn-Mutationen tragen, die zur Entwicklung einer AML prädisponieren. Derartige Keimbahn-Mutationen werden nicht nur bei kindlichen Leukämien, sondern in zunehmendem Maße auch bei Leukämien im Erwachsenenalter identifiziert.
Krankheitszeichen
Die Symptome der AML werden im Wesentlich durch die Verdrängung der normalen Blutbildung bestimmt:
- Anämie (Blutarmut): Durch eine Abnahme der Sauerstoffträger kommt es zu Müdigkeit, Blässe, Atemnot und Abgeschlagenheit
- Leukopenie und Neutropenie (Abnahme der Immunzellen): Infektionen durch Bakterien, Pilze und Viren, die teilweise einen sehr schweren Verlauf haben
- Thrombopenie (Abnahme der Blutplättchen): Blutungen, die bei Befall lebenswichtiger Organe wie das Gehirn lebensbedrohlich werden können
Die Infiltration von extramedullären Organen und Geweben wie Leber, Milz, Lymphknoten, Knochen, Gingiva (Zahnfleisch), Haut und zentralem Nervensystem kann unterschiedliche Symptome und Befunde zur Folge haben:
- Hepatomegalie (Lebervergrößerung)
- Splenomegalie (Milzvergrößerung)
- Lymphadenopathie (Lymphknotenschwellungen)
- Knochenschmerzen
- Gingivahyperplasie (Zahnfleischschwellung)
- Unspezifische neurologische Symptome (z.B. Kopfschmerzen, Schwindel, Seh- und Gefühlsstörungen)
Eine tumorbildende Leukämiemanifestation außerhalb des Knochenmarks wird als Chlorom bezeichnet.
Sehr hohe Leukozytenzahlen können zu Symptomen der Leukostase (Sehstörungen, zentralnervöse Ausfallserscheinungen, Blutungsereignissen) führen. Bei den Laboruntersuchungen findet sich häufig eine Erhöhung der Laktatdehydrogenase, seltener sind Elektrolytstörungen (Hyper-/Hypokaliämie) oder eine Hyperurikämie nachweisbar. Gerinnungsstörungen finden sich vor allem bei der akuten Promyelozytenleukämie (APL) in Form eines disseminierten intravasalen Gerinnungssyndroms (DIC).
Diagnostik
Zum Standard der Diagnostik gehören die morphologische Begutachtung des Blut- und Knochenmarkausstriches, die sogenannte Immunphänotypisierung (mittels Durchflusszytometrie), sowie die zytogenetische und molekulargenetische Analyse.
Im Blut- und Knochenmarkausstrich zeigt sich charakteristischerweise eine Vermehrung myeloischer Blasten über 10-20% (im normalen Knochenmark weniger als 5%). Bei der Immunphänotypisierung werden mit Hilfe eines Panels von monoklonalen Antikörpern spezifische Moleküle auf der Oberfläche und im Inneren der Leukämiezellen nachgewiesen. Die Immunphänotypisierung dient der Bestimmung der Linienzugehörigkeit (AML versus andere Leukämieformen) sowie der Identifizierung eines Leukämie-assoziierten Phänotyps (LAIP), den man im Behandlungsverlauf für den Nachweis minimaler Resterkrankung verwenden kann.
Klassifikation
Die Klassifikation der Erkrankung erfolgt nach der aktuellen International Consensus Classification (ICC). Diese berücksichtigt ganz überwiegend die zugrundeliegenden genetischen Veränderungen.

Therapeutische Möglichkeiten
Patienten, die für eine intensive Chemotherapie eligibel / geeignet sind
Die Behandlung der AML basiert nach wie vor auf der Gabe einer intensiven Chemotherapie, wobei Zytostatika (Medikamente mit hemmender Wirkung auf die Zellteilung) mit unterschiedlichen Wirkmechanismen kombiniert werden. Im Wesentlichen werden hier zwei Zytostatika, die Anthrazykline und Cytosin-Arabinosid (Cytarabin, Ara-C) eingesetzt, die sich seit Jahren in der Behandlung der AML bewährt haben.
Für bestimmte Hochrisiko-Formen der AML wurde CPX-351 (Vyxeos®) von der Food and Drug Administration (FDA) und der European Medicines Agency (EMA) zugelassen. Bei CPX-351 handelt es sich um eine liposomale Verpackung der beiden Zytostatika Cytarabin und Dunorubicin.
Für die AML mit FLT3 Mutationen wurde der FLT3 Inhibitor (Hemmstoff) Midostaurin (Rydapt®) in Kombination mit intensiver Chemotherapie zugelassen.
Im Rahmen von klinischen Studien werden weitere, vielversprechende neue Medikamente getestet mit dem Ziel die Behandlungsergebnisse immer weiter zu verbessern. Zu diesen Medikamenten gehören beispielsweise der BCL-2 Inhibitor Venetoclax und die IDH1- und IDH2-Inhibitoren Ivosidenib bzw. Enasidenib. Alle diese Substanzen können wir unseren Patienten im Rahmen von kontrollierten Therapiestudien anbieten.
Verlauf der Behandlung: Bei der Behandlung der AML muss zunächst durch eine erste Chemotherapie, die sogenannte Induktions-Chemotherapie, eine Remission der Erkrankung erreicht werden. Unter einer vollständigen Remission versteht man die mikroskopisch nachweisbare Reduktion der Leukämiezellen („Blasten“) im Knochenmark unter 5% bei gleichzeitiger Normalisierung der Blutbildung. Nach einer Induktions-Chemotherapie ist es zunächst möglich, bei ca. 70-80% der Patienten im Alter zwischen 18 und 60 Jahren eine Remission der Erkrankung zu erzielen. Ohne weitere chemotherapeutische Behandlung ist allerdings das Risiko sehr hoch, dass nach kurzer Zeit die Leukämie zurückkehrt (Rezidiv). Deshalb schließt sich an diese Induktions-Therapie die Konsolidierungs-Chemotherapie an mit dem Ziel, die noch verbleibenden Leukämiezellen zu töten und eine dauerhafte Leukämiefreiheit zu sichern. Trotz intensiver Konsolidierungs-Chemotherapien erleiden ca. 30-40% der Patienten mit AML ein Rezidiv. Nach einem Rezidiv sind die Erfolgsaussichten auf eine erneute Chemotherapie eine Remission zu erreichen geringer. Deshalb ist es von entscheidender Bedeutung, ein Rezidiv der Erkrankung zu verhindern. Seit einigen Jahren ist bekannt, dass bestimmte genetische Veränderungen mit höherer bzw. niedrigerer Rezidiv-Wahrscheinlichkeit vergesellschaftet sind. Eine umfassende genetische Analyse bei Diagnosestellung kann somit zur Abschätzung des Rezidiv-Risikos beitragen. Bei Patienten, die aufgrund der vorliegenden genetischen Veränderungen ein hohes Rezidiv-Risiko aufweisen, empfehlen wir die Durchführung einer allogenen Blutstammzell-Transplantation. Die Blutstammzell-Transplantation ist das Verfahren, das am wirkungsvollsten das Risiko eines Rezidivs verhindern kann, dieses ist jedoch mit einer höheren Therapie-assoziierten Sterblichkeit vergesellschaftet (s.u.).
Bei der akuten Promyelozyten-Leukämie (APL) handelt e sich um eine seltene Form der AML, die grundsätzlich einer anderen Therapie bedarf. Die APL ist durch die chromsomale Translokation t(15;17) mit daraus resultierender PML-RARA Gen-Fusion gekennzeichnet. Die Therapie dieser Leukämie form besteht aus der Kombinationsbehandlung mit dem Vitamin-A-Derivat All-trans Retinsäure (ATRA) und Arsentrioxid. Mit dieser „Chemotherapie-freien‘ Kombinationsbehandlung kann diese Leukämieform heute in über 90% der Fälle geheilt werden.
Ältere Patienten, die für eine intensive Chemotherapie nicht eligibel / geeignet sind
Die Therapieergebnisse bei älteren Patienten (>65-70 Jahre) sind deutlich schlechter als die bei jüngeren Patienten. Die Rate an kompletten Remissionen - also eine Normalisierung des Blutbildes und Beseitigung der Blasten im Knochenmark (<5%) - liegt bei älteren Patienten zwischen 50% und 70%. Ursächlich sind einerseits Patienten-bezogene Faktoren, d.h. Begleiterkrankungen wie z.B. Bluthochdruck, Diabetes mellitus, die zu Komplikationen führen, andererseits das Vorhandensein von prognostisch ungünstigen genetischen Faktoren.
In Abhängigkeit von der genetischen Subgruppe der Leukämie und den Begleiterkrankungen gibt es verschiedene Therapieformen, die sich im Hinblick auf Toxizität und Wirkung unterscheiden. Mit herkömmlicher Chemotherapie werden nur bei ca. 10-15% der Patienten Langzeitremissionen erreicht. Auch bei älteren Patienten ist prinzipiell eine allogene Stammzelltransplantation möglich, wobei hier der Allgemeinzustand des Patienten sowie die vorhandenen Begleiterkrankungen von entscheidender Bedeutung sind.
Für Patienten, die für eine konventionelle intensive Chemotherapie nicht geeignet sind, ist die Kombination einer sog. hypomethylierenden Substanzen (Azacytidin, Decitabin) mit dem BCL-2 Inhibitor Venetoclax (VENCLYXTO®) die Therapie der Wahl. Mit diesen Therapien können Remission in ca. 60% der Fälle erzielt werden, die mediane Lebenserwartung liegt jedoch nur bei ca. 12-15 Monaten. Zahlreiche neue Therapie-Kombinationen sind in der klinischen Prüfung.
Allogene Stammzelltransplantation
Die allogene Knochenmark- oder Blutstammzell-Transplantation (allogene SZT) stellt für viele jüngere, aber mittlerweile auch viel ältere AML-Patienten, insbesondere für diejenigen, bei denen eine Hoch-Risiko-Konstellation besteht, die derzeit einzige kurative Therapieoption dar. Die allogene SZT ist ein Behandlungsverfahren, das an einem hochspezialisierten Zentrum durchgeführt werden sollte, welches über ein Team von Ärzten und Pflegepersonal mit langjähriger Expertise verfügt sowie die notwendige Infrastruktur besitzt.
Das allogenen SZT hat zwei therapeutische Prinzipien. Zum einen erhält der Patient eine intensive Chemotherapie (ggf. in Kombination mit einer Ganzkörper-Strahlentherapie), die zur Abtötung der Leukämien führen soll. Zum anderen entsteht durch die Infusion des Spender-Transplantats eine Immunreaktion der Spender-Immunzellen gegen die noch verbliebenen Leukämiezellen im Empfänger (sog. Graft versus Leukemia Effekt). Die unerwünschte Reaktion und somit das Risiko der allogenen SZT besteht vor allem darin, dass die Spender-Immunzellen nicht nur gegen die Leukämiezellen wirken, sondern eine Immun-Reaktion auch gegen andere Organe auslösen (z.B. Haut, Leber, Magen-Darm; sog. Graft versus Host Disease). Diese GvHD-Reaktion ist die Haupt-Ursache für die Therapie-assoziierte Sterblichkeit bei der allogenen SZT, die zwischen 10% und 25% liegt.
Bei der allogenen SZT werden Stammzellen von einem geeigneten Spender auf den Empfänger übertragen. Die Eignung eines Spenders wird durch eine Blutuntersuchung, die sogenannte HLA-Typisierung, festgestellt. Als Stammzellspender werden entweder HLA-identische Familienspender (insbesondere Geschwister) oder, sofern kein passender Familienspender zur Verfügung steht, ein HLA-verträglicher Fremdspender verwendet. In den verschiedenen Spenderdateien stehen weltweit mehr als 10 Millionen Fremdspender zur Verfügung. Derzeit gelingt es für 90% aller Patienten innerhalb von 1-3 Monaten einen passenden Fremdspender zu finden.
Förderung der AML-Forschung an der Universitätsklinik Ulm
- Biomedizinische und Pharmazeutische Industrie
- Deutsche Forschungsgemeinschaft (DFG), u.a. Sonderforschungsbereich 1074
- Bundesministerium für Bildung und Forschung (BMBF)
- Deutsche Krebshilfe
- Europäische Union
- Baustein- und Clinician Scientist Program, Medizinische Fakultät der Universität Ulm
- Else Kröner-Fresenius-Stiftung
Stand: 5.2.2023